Is There an Automatic “Calculator” for Molecular Orbitals?
Yes, several software tools can automatically calculate and visualize molecular orbitals and their diagrams, but no simple single-click calculator instantly produces full, detailed molecular orbital drawings without user input. Instead, users rely on a series of programs for building molecular structures, computing orbitals, and visualizing results. These tools range from free and open-source to commercial packages requiring licenses.
Software Categories for Molecular Orbital (MO) Calculations

The molecular orbital calculation and visualization landscape includes:
- Commercial Quantum Chemistry Programs: Such as Spartan, Gaussian, GAMESS, ORCA, MOPEG. These provide comprehensive, high-accuracy calculations but usually require licenses or institutional access.
- Free and Open-Source Tools: Including IBOView, IQmol, Avogadro, ChemCompute, ChemCraft. These offer various degrees of functionality, often combining building, calculation, and visualization.
- Supporting Visualization and Educational Resources: Websites like ptable.com offer static MO diagrams for elements; simple searches often yield diagrams for common molecules.
Workflow for Automatic Molecular Orbital Calculation and Visualization

- Build 3D Molecular Structure: Use molecule-building software such as IQmol or Avogadro. These tools allow users to draw molecules and optimize their geometries.
- Save Molecular Geometry: Export the structure in formats like XYZ files that store atomic coordinates.
- Compute Molecular Orbitals: Use software such as IBOView, GAMESS (often via ChemCompute online platform), or other quantum packages to perform calculations.
- Visualize Orbitals: Many programs display orbital shapes directly. ChemCraft and Atom app can be used for further visualization and interpretation.
This multi-step process technically automates calculation and visualization but does not offer a fully integrated, instant MO diagram drawing tool commonly found in textbooks.
Free and Open-Source Options for Molecular Orbitals

- IBOView: A user-friendly program focused on localized molecular orbitals based on Density Functional Theory (DFT). It does not have molecular building features, so external programs like Avogadro are needed.
- IQmol and Avogadro: Both provide molecule-building interfaces with rudimentary optimization and the option to export structures for orbital calculation.
- ChemCompute: A web-based platform primarily using GAMESS for calculations, suitable for students and researchers without local software installations.
- ChemCraft: Useful for visualizing and interpreting orbitals post-calculation, often accessible via university licenses.
- Atom App: Mentioned as an application capable of calculating orbitals, though less widely detailed in references.
Limitations and Conceptual Considerations

Molecular orbitals computed via software represent complex quantum states. Unlike simplified textbook models, real calculated orbitals are delocalized and can involve mixing orbitals over multiple atoms.
Programs like IBOView attempt to localize these orbitals to resemble classical bonding concepts, but the output will often still look different from idealized images used in teaching.
Fully automatic MO calculators that draw textbook-style molecular orbital diagrams including energy levels and orbital interactions are not commonly available as standalone tools. Expertise is typically required to generate and interpret these results.
Additional Tools and Programming Approaches

For advanced users, Python environments with Anaconda and visualization libraries can help simulate and animate molecular orbitals with interactive controls. This approach requires programming knowledge but offers flexibility for research and education.
Summary Table of Tools for Molecular Orbital Calculations

| Tool | Function | License | Notes |
|---|---|---|---|
| Spartan | Comprehensive MO calculations and visualization | Commercial | High cost, full features |
| GAMESS / Gaussian / ORCA | Quantum chemical computations | Mostly free (GAMESS, ORCA), Gaussian commercial | Widely used in research |
| IBOView | Orbital visualization with localization focus | Free/Open-source | Needs external molecule builder |
| IQmol / Avogadro | Molecule builder with optimization | Free/Open-source | Export to calculation programs |
| ChemCompute | Online calculation portal (GAMESS backend) | Free | Accessible to students |
| ChemCraft | Visualization and analysis of orbitals | Free or license via institution | User friendly |
Key Takeaways
- No single, fully automatic calculator produces textbook-style molecular orbital diagrams instantly.
- Users combine molecule-building, computing, and visualization tools for full MO analysis.
- Commercial packages provide powerful features but require licenses.
- Several free tools like IBOView, IQmol, Avogadro, and ChemCompute facilitate MO calculations and viewing.
- Calculated orbitals are more complex than simplified classroom diagrams; localization techniques help make them intuitive.
- Programming via Python and Anaconda offers customizable MO visualization for advanced users.
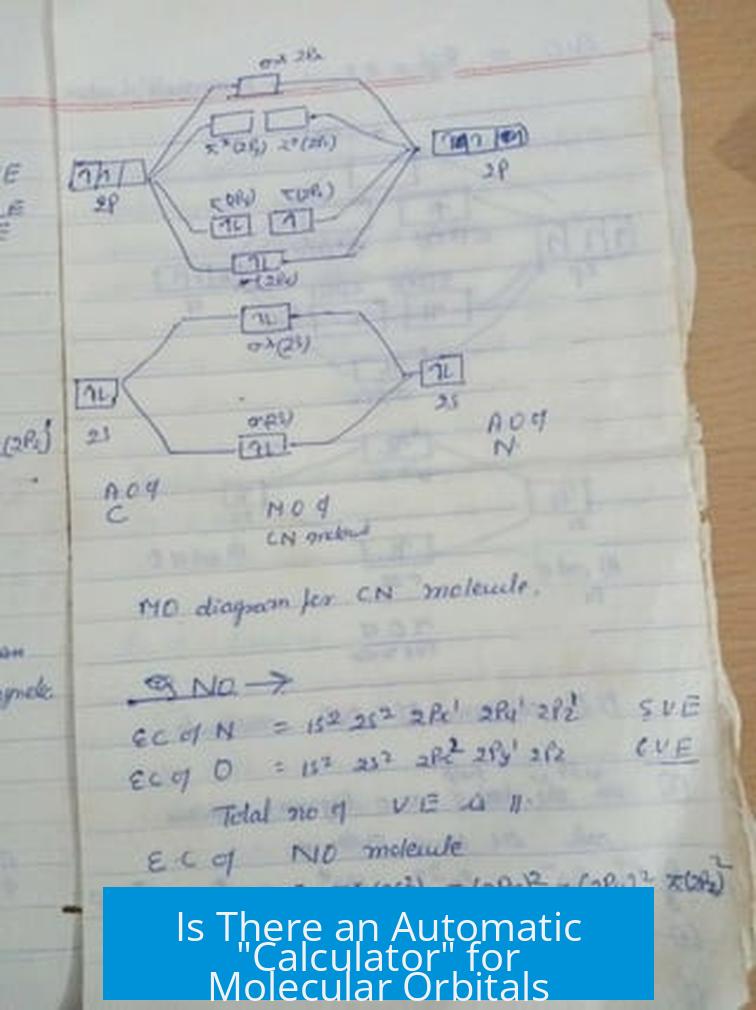
Is There an Automatic “Calculator” for Molecular Orbitals (the Drawing with All the Orbitals)?
Short answer: There’s no magic one-click molecular orbital (MO) calculator that instantly churns out those neat, full orbital diagrams as you might wish. Instead, you’ll use a smart cocktail of molecule building tools, quantum software, and visualization apps to get there.
Now, that might sound like a bummer if you hoped for something as simple as “click-and-draw MO.” But stick with me. This journey has multiple stops, and each one is packed with practical tips, tools, and some honest realities.
Why Isn’t There a Single Automatic MO Diagram Calculator?
Fundamentally, molecular orbitals come from complex quantum mechanical calculations. The “automatic” part means software has to solve mathematical models that can require heavy computational power and expertise. The visual results are also often more intricate than the neat, textbook drawings we learn in class.
The classroom MO diagrams are simplified models — elegant and useful for understanding how atomic orbitals mix. Actual computed orbitals, however, blend and delocalize across atoms in ways that aren’t always straightforward to interpret or depict clearly. So, a simple “automatic” tool that instantly produces a full MO drawing accessible to everyone doesn’t really exist.
How Do Scientists and Students Normally Generate MO Diagrams?
The typical workflow involves:
- Building your molecule’s 3D structure using intuitive tools like Avogadro or IQmol. These programs let you sketch molecules in 3D and clean up messy structures with basic optimization.
- Saving the molecular geometry as a file (often XYZ format) for further processing.
- Running MO calculations with quantum chemistry software such as IBOView (free and user-friendly for viewing orbitals) or more powerful packages like GAMESS, Gaussian, or Spartan. Note that most advanced tools need licenses or institutional access.
- Visualizing the results, sometimes in the same software (like IBOView) or using dedicated visualization tools such as ChemCraft or the Atom app.
Want an easy example? Use Avogadro to build your molecule, export it, then load it into IBOView to see molecular orbitals rendered based on density functional theory (DFT). This setup is about as automatic as it gets for free tools.
Let’s Talk About the Tools: What’s Out There and What You Need to Know
On the paid side, Spartan, Gaussian, GAMESS, and Orca stand out. They offer deep, precise computations and superb visualization, but licenses can be costly and usually require institutional support. These are the “heavy artillery” of computational chemistry software.
If you’re on a budget or just curious, there are excellent free alternatives:
- IBOView: Its main gig is *localizing* molecular orbitals to look more like the classic orbitals you remember from school. It’s simple, lightning fast, and based on DFT. However, it can’t build molecules — you’ll have to provide structures from elsewhere.
- Avogadro and IQmol: Both act as molecule builders and optimize structures a bit. IQmol sometimes integrates with GAMESS and can do some orbital visualization, making it a neat combo.
- ChemCompute: It’s a free web platform that runs GAMESS computations and guides newcomers gently through MO calculations. Perfect if you’re still getting comfortable with computational chemistry jargon.
- ChemCraft and Atom app: Handy visualization tools for viewing orbital/hybridization results. ChemCraft is often free or available through universities.
Pro TIP: For best results, combine software — build the molecule in Avogadro, compute orbitals in IBOView or GAMESS (via ChemCompute), then visualize with ChemCraft or Atom app.
Why Do the Computed Orbitals Look Different from Classroom Diagrams?
Think of your classroom MO diagrams as friendly cartoons—they’re simplified sketches emphasizing conceptual points. Real MO calculations reveal that orbitals can spread (delocalize) across the entire molecule, especially in complex compounds. This can look confusing at first.
IBOView’s main purpose is to *localize* these orbitals to mimic the shapes and types taught in class. Even so, expect surprises. The software’s orbital shapes may not always align perfectly with your textbook pictures, but they’re scientifically more accurate.
Can You Get Molecular Orbital Visuals Without Complex Software?
Sometimes yes, sometimes no. For quick looks, Google image search can be your best friend—search “molecular orbital diagram [molecule name]” and find ready-made pictures for small simple molecules.
Also, websites like ptable.com offer small MO figures for elements, which helps build intuition.
But if your goal is precise, customizable diagrams for novel molecules, you’ll want software tools after all.
Looking for Something Programmable and Customizable?
If you have a coding mindset, installing Python through Anaconda with visualization libraries can offer an interactive playground. You can tweak geometries or orbitals with sliders and run simulations. Though this requires some programming savvy, it opens doors to advanced customizations.
Practical Example: How to Get a Molecular Orbital Diagram Automatically (Sort Of)
- Open Avogadro. Sketch your molecule (e.g., benzene).
- Optimize the structure using Avogadro’s built-in functions.
- Export the molecule as an XYZ file.
- Load that XYZ file into IBOView.
- Run the orbital calculation (based on DFT) within IBOView.
- View the orbitals—it’s not a simple diagram filled with energy levels and arrows, but you get full 3D renderings of HOMO, LUMO, and others.
- Optionally export images or manipulate orbitals with visualization software like ChemCraft.
Voilà—almost automatic, and based entirely on free tools.
Final Thoughts and Recommendations
Is there an all-in-one, foolproof automatic calculator that draws textbook-style complete molecular orbital diagrams? Not really. The field requires a combination of software pieces working hand-in-hand.
If you’re a student or beginner, I strongly recommend dipping toes with Avogadro and IBOView. They minimize technical headaches while delivering meaningful orbital visuals. For deeper dives, explore ChemCompute on the web or, if accessible, Spartan or Gaussian.
Wondering where to download everything or how to get started? Here’s the quick scoop:
- Avogadro: Free molecule builder.
- IBOView: Quick orbital calculations and visualization.
- IQmol: Another builder/visualizer combo.
- ChemCompute: Web-based GAMESS interface.
- Ptable: Molecular orbital icons for elements.
Before you dive in, a question to ponder: How much detail do you really need in your MO diagram? Sometimes, a simple sketch helps more than a full quantum-mechanical model tangled with delocalizations. But if you crave accuracy and real-world data, the right tools will get you there—with a bit of patience and curiosity.
Now that you know the landscape, why not try building your first molecule today? The world of molecular orbitals is richer and more fascinating once you see it in 3D.
Is there an automatic tool that instantly draws all molecular orbitals for a molecule?
No single tool provides instant full molecular orbital diagrams automatically. Most workflows combine molecule builders, orbital calculators, and visualization tools to generate and view orbitals.
Which free software can I use to calculate and visualize molecular orbitals?
IBOView offers a simple orbital calculator based on DFT. You need a molecule builder like IQmol or Avogadro to create structures. ChemCompute uses GAMESS for calculations and is free.
Why do calculated molecular orbitals look different from those in textbooks?
Textbook orbitals are simplified models. Real molecular orbitals are delocalized and complex. Software like IBOView tries to localize orbitals closer to textbook examples, but differences remain.
Can I build molecules and calculate orbitals without buying expensive software?
Yes. Use free molecule builders like Avogadro or IQmol, then calculate orbitals with free tools like IBOView or online platforms such as ChemCompute, which runs GAMESS calculations for free.
Are there visualization tools for exploring molecular orbitals without deep computational chemistry knowledge?
ChemCraft and the Atom app let you view orbitals easily. Also, Python with Anaconda and visualization add-ons provide interactive views but require some programming skills.

Leave a Comment