Troubleshooting Cloning: Ensuring Successful DNA Transformation

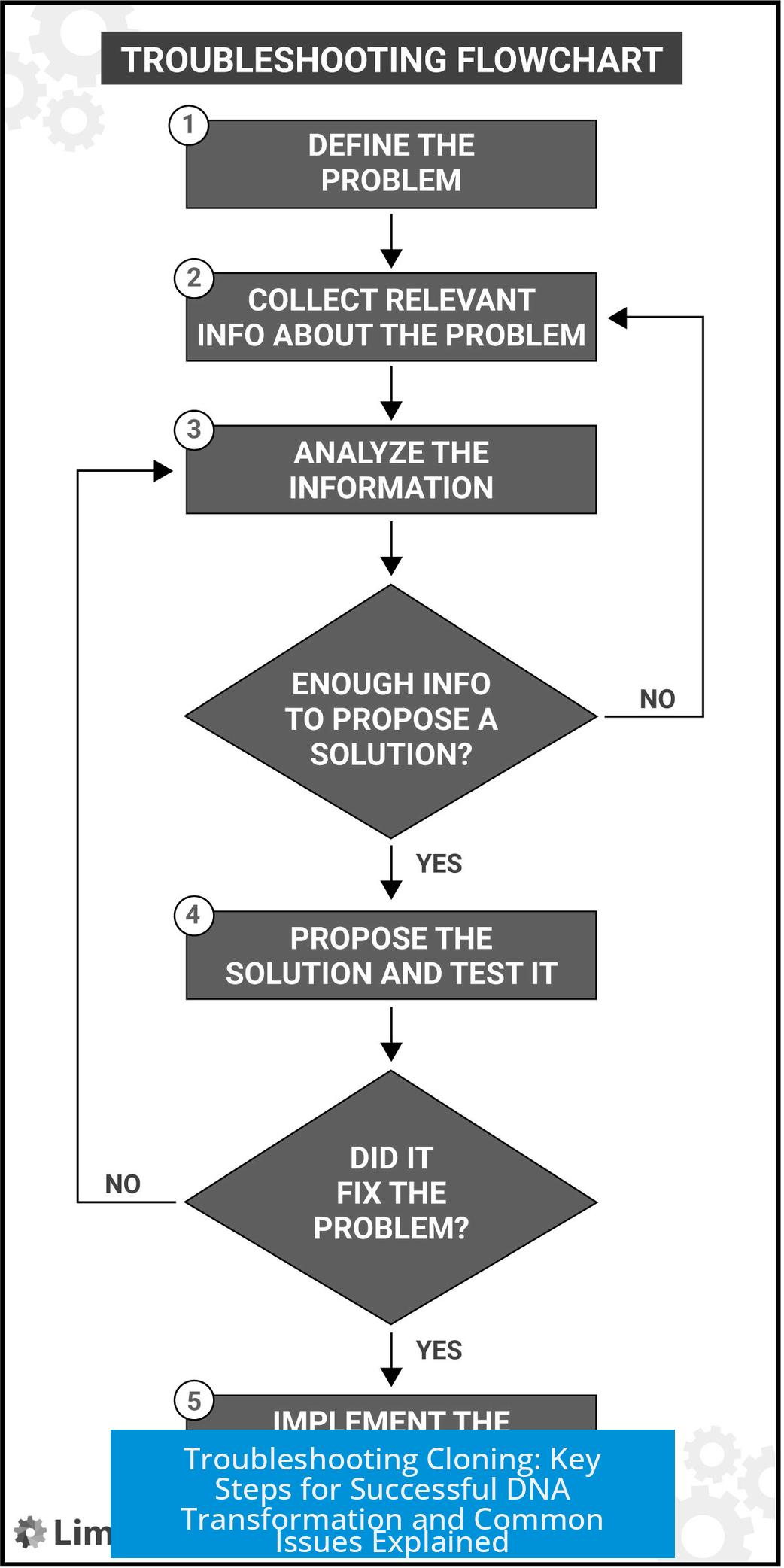
Troubleshooting cloning primarily involves optimizing competent cell quality, transformation protocols, and plasmid integrity. Addressing each element systematically can identify issues and improve colony yields significantly.
Competent Cells: Quality and Preparation
Competent cells are crucial for cloning success. Their ability to take up foreign DNA defines the transformation efficiency.
Methods of Preparing Competent Cells

- Commercially available competent cells often offer consistent high efficiency.
- Homemade cells are frequently prepared via the rubidium chloride method, shown to outperform the calcium chloride method in terms of transformation success, especially with challenging ligations, such as PCR products.
- The calcium chloride approach often fails with complex ligations and yields few if any colonies.
- Proper handling includes keeping all materials on ice during preparation and snap freezing aliquots with approximately 15% glycerol to maintain viability.
- Aliquots of 200 μL are common, with 60 μL used per transformation.
- Freezing and thawing cycles should be limited to avoid damaging cells and reducing competency.
- Seed stock freshness is vital; old or tired seed stocks can drastically lower efficiency.
- Using the appropriate competent strain is important; for example, DH5α is not suitable for all plasmids—some require strains like Sbl3.
Handling During Transformation
- Post-heat-shock recovery involves adding 150 μL medium to 60 μL of cells and shaking at 180 rpm for 1 hour at 37°C for optimal recovery.
- Plating strategies include either spreading cells with glass beads or a spreader; using multiple plates increases chances of isolating single colonies.
- Positive controls such as pUC19 plasmid should be transformed alongside experimental samples to verify cell competence.
Transformation Protocol Details

Heat Shock and Electroporation
- Heat shock temperature control is critical. Use a water bath to maintain a consistent temperature, often 42°C for 30-60 seconds, to improve efficiency.
- Electroporation requires optimizing the time constant and fast handling; swift addition of DNA immediately upon thawing and prompt pulsing increase the transformation rate.
- Water bath heat shock often outperforms other methods according to practical lab observations.
Recovery Medium

- While LB can substitute in some cases, SOC medium is generally preferred for the recovery step after transformation; it contains nutrients and components that aid cell recovery after the stress caused by heat shock or electroporation.
- Using nutrient-rich SOC or similar media can significantly increase transformation efficiency, particularly for ligation-derived plasmids.
- Protocols optimized for specific cell and vector combinations often differ; what works in one lab might not translate directly to another.
Plasmid and Cloning Strategy
Plasmid Quality and Verification
- Low colony counts from transformations with uncut plasmid suggest poor plasmid quality or compromised cells.
- Transforming a confirmed, high-quality plasmid from another lab member can help distinguish plasmid versus cell issues.
- Sequencing your vector pre- and post-transformation ensures plasmid identity and integrity.
- Plasmids showing unexpected sequences or variations may require replacing with a freshly prepared or newly sourced plasmid.
Cloning Methods to Increase Efficiency
- Traditional restriction enzyme digestion with ligation is common but sometimes inefficient, especially for PCR-derived fragments.
- Alternative approaches like TA cloning for PCR products provide easy ligation compatible ends.
- Modern cloning methods such as Gibson Assembly, NEBuilder, or Infusion cloning offer one-pot solutions combining PCR-amplified inserts with linearized vectors bearing homologous overlaps.
- Workflow for Gibson assembly typically includes:
- Inverse PCR of vector with 15 bp overlaps matching the insert
- PCR amplification of insert with complementary overlaps
- Assembly reaction at 50°C for 20 minutes
- Transformation into competent cells
- Golden Gate Assembly uses type IIS restriction enzymes for scarless, directional assembly, offering alternative high-efficiency cloning.
- These techniques reduce background colonies by selectively amplifying the desired recombined plasmid.
PCR Product Purification
- Gel extraction of PCR products is common but can damage DNA due to UV exposure or introduce impurities.
- Using PCR cleanup kits avoids UV damage and yields clean fragments appropriate for cloning.
- Digestion with DpnI removes methylated template DNA leftover from PCR, enriching the preparation for newly amplified inserts.
Additional Troubleshooting Tips
Environmental and Procedural Controls
- Always verify the pH of reagents, as variations can affect enzyme activity or transformation efficiency.
- Minimize UV exposure during gel electrophoresis and select the lowest power, longest wavelength settings available to reduce DNA damage.
- Monitoring transformation efficiency in parallel with positive controls helps determine whether issues lie with cells, plasmids, or the protocol.
- Observing someone else’s successful transformation can serve as a benchmark and help identify procedural differences.
Summary of Key Troubleshooting Steps
- Use high-quality competent cells prepared with optimized methods like rubidium chloride and minimize freeze-thaw cycles.
- Employ consistent heat shock with a water bath or optimize electroporation parameters for rapid and efficient DNA uptake.
- Prefer SOC or similarly enriched recovery media to enhance cell recovery post-transformation.
- Confirm plasmid identity and quality by sequencing and control transformations with known vectors.
- Consider modern cloning techniques (Gibson Assembly, Golden Gate) to improve efficiency and reduce background colonies.
- Avoid damaging PCR product purification methods and use cleanup kits with DpnI digestion where appropriate.
- Maintain careful control of reagent pH and minimize UV exposure to protect DNA integrity.
Troubleshooting Cloning: Why Isn’t My Transformation Working?
Cloning can be tricky, but if you’re pulling your hair out over low transformation efficiency or no colonies, the root cause is usually your competent cells or the procedure around handling them. Let’s dive into the nitty-gritty of troubleshooting cloning with specific tips about competent cells, the transformation protocol, plasmid quality, and little tricks to boost your success rate.
Competent Cells: The Heart of Successful Cloning
Imagine competent cells as the welcoming committee for your plasmid DNA—they make or break your cloning experiment. If your cells aren’t “competent” enough, you get poor or no colonies.
How prepared are your cells? Are you using commercial competent cells or making them yourself? The method you choose hugely impacts success. For self-prep, the rubidium chloride method reigns supreme. It outperforms the inferior calcium chloride method, which often yields zero colonies on tricky ligations like PCR products. Talk about frustration!
Worried about the recovery medium? Some swear by adding SOC after heat shock—nutrient-rich and designed to help bacterial cells bounce back after trauma. But others find LB medium does the job just as well. The consensus? Your cells’ quality matters far more than the medium you use.
- Always keep cells on ice during preparation. This chilling restraint keeps everything stable and crisp.
- Snap freeze in aliquots (~200 μl) with 15% glycerol. This prevents repeated freeze-thaw cycles—a major killer of competence.
- Use seed stocks fresh from a trusted source. Old or tired stocks tank efficiency dramatically.
- Limit freeze-thaw and handle the aliquots carefully during transformation.
- Test your cells with a positive control plasmid like pUC19. No colonies? Definitely the cells’ fault.
Remember: competent cells exposed to ~100 ng of a fully assembled plasmid should give a thick lawn of colonies in just 5 minutes. If not, they’re just not competent enough.
Transformation Protocol: Timing and Temperature Are Key
Are you confident your heat shock step is spot-on? Heat shock timing, and critically, temperature accuracy, cannot be overstated. Use a water bath—not a dry block—for best results. Many labs find water baths make transformations zing along better, though the exact reason remains a bit of a mystery.
For electroporators, check the time constant carefully. An incorrect pulse can fry your cells or yield almost no transformants.
Act fast! As soon as the cells thaw, add your DNA immediately, then heat shock or electroporate without delay. Speed matters.
Post shock, recovery is another crucial stage:
- Add ~150 μl recovery medium to your 60 μl heat-shocked cells.
- Shake rapidly (180 rpm) at 37°C for about an hour in tubes like Eppendorf tubes.
- Plate the whole mixture onto a selective agar plate.
- If you want single colonies, use glass beads or a sterile “hockey stick” spreader to dilute your cells across multiple plates.
Plasmid and Cloning Strategy: Don’t Overlook the DNA
Before blaming your cells or protocol, verify your plasmid backbone or insert quality. Alarm bells should ring if your uncut plasmid produces low colony counts.
Try co-transforming a known “good” plasmid from a trusted colleague. If that also yields poor colonies, your competent cells or protocol is suspect. But if the control does well and yours doesn’t, “game over”—your plasmid may be defective or the prep low quality.
Sequence your vector. It’s boring but essential. Errors or contamination in the plasmid prep can stealthily wreck transformations. If your vector sequences don’t match expectations, get a fresh prep—preferably from a different source.
Consider Alternative Cloning Approaches to Boost Efficiency
Are you stuck with classical ligation? Maybe it’s time to upgrade. Methods like Gibson Assembly or Golden Gate Assembly are your friends.
- Gibson Assembly: Join DNA fragments with overlapping ends in a one-pot 50°C reaction for 20 minutes. This method reduces reliance on perfect ligation efficiency and generally yields higher cloning success.
- Golden Gate Assembly: Uses Type IIS restriction enzymes for seamless multi-fragment assembly in one reaction.
- If you do PCR amplify your insert, clean the PCR products using a cleanup kit rather than gel extraction to avoid DNA damage. Adding DpnI digestion helps remove methylated template DNA from PCR reactions, improving the final insert quality.
Inverse PCR with overlapping primers can generate inserts with built-in homology arms aligning specifically with your vector, reducing background colonies from uncut or incorrect plasmids. This technique also indirectly confirms your vector identity by amplifying just the desired plasmid version.
Extra Tips to Avoid Hidden Dead Ends
- Check pH of all buffers used in cloning reactions. A pH off by just a little can kill enzyme efficiency.
- Protect DNA from overexposure to UV during gel electrophoresis by using the lowest UV power and longest wavelength. Short-wavelength UV zaps your cloning DNA.
- Shadow an experienced colleague performing transformations with a standard plasmid. Compare colony numbers—this real-time check clears up if your cells or protocol need tuning.
- Limit freeze-thaw cycles. Competent cells are delicate; repeated thawing slashes efficiency.
So, What’s the Takeaway?
Your cloning problems usually boil down to one of three culprits: competent cells of subpar quality, DNA of poor quality or incorrectly verified identity, and procedural slips during transformation.
Start with fresh, well-prepped, properly frozen competent cells made by the rubidium chloride method or trusted commercial stocks. Use a positive control plasmid, verify your vector by sequencing, and consider advanced cloning methods like Gibson Assembly for tough inserts.
Pay close attention to your transformation protocol. Use a water bath set at correct temperature for heat shock, add DNA quickly after thawing, shake recovery cultures properly, and plate carefully to get discrete colonies.
And remember to keep buffers and reagents fresh and properly pH-adjusted while protecting DNA from harmful UV exposure.
Final Thought
If cloning was simple, it wouldn’t be called “cutting-edge” molecular biology, right? Don’t be shy about troubleshooting methodically and logging every step. Your cloning success will improve dramatically if you treat the process as a detective mystery where every clue counts.
Feeling stuck with your current protocol? Take a step back, check cells, sequence your plasmid, and maybe try that Gibson Assembly everyone’s buzzing about. Soon enough, you’ll be eating up those transformants and moving on to exciting experiments instead of troubleshooting forever.
Q1: How can I tell if my competent cells are the cause of poor cloning efficiency?
If your competent cells produce few or no colonies even with a control plasmid, they’re likely inefficient. Competent cells should show many colonies after a short heat shock with a known good plasmid.
Q2: What recovery medium should I use after transformation for best results?
Using SOC or other nutrient-rich broth helps cells recover after heat shock. While LB might work sometimes, richer media improve transformation efficiency, especially for tricky cloning reactions.
Q3: Why might my uncut plasmid yield very low colony counts?
This can indicate plasmid issues. Try transforming a verified plasmid alongside yours. Also, sequence your vector and any colonies from vector-only transformation to confirm it matches the expected sequence.
Q4: Are there cloning methods that improve transformation success over traditional ligation?
Methods like Gibson Assembly or Golden Gate Assembly often yield higher efficiencies. They use one-pot reactions and reduce background by amplifying the correct construct, helping especially if your vector prep is not pure.
Q5: What is the best way to prepare competent cells for cloning?
Use fresh seed stocks and prepare cells using rubidium chloride method. Keep everything cold on ice, snap freeze aliquots with glycerol, and avoid multiple freeze-thaw cycles to maintain high competency.


Leave a Comment